Substructure and similarity searching a large dataset#
Open Babel provides a format called the fs -- fastsearch index which should be used when searching large datasets (like ChEMBL) for molecules similar to a particular query. There are faster ways of searching (like using a chemical database) but FastSearch is convenient, and should give reasonable performance for most people.
To demonstrate similarity searching, we will use the first 1000 molecules in the latest release of ChEMBL:
Download the 2D SDF version of ChEMBL,
chembl_nn.sdf.gz, from the ChEMBLdb download site and save in your Work folder. (Note: this is a gzipped file, but Open Babel will handle this without problems.)Set up an SDF to SDF conversion, set
chembl_nn.sdf.gzas the input file and1000_chembl.sdfas the output file.Only convert the first 1000 molecules by entering
1000in the box End import at molecule # specified.

Click CONVERT
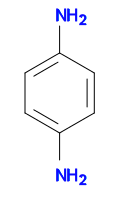
We can going to use the following structure for substructure and similarity searching. It can be represented by the SMILES string Nc1ccc(N)cc1.
Next, we will create a FastSearch index for this dataset of 1000 molecules:
Convert
1000_chembl.sdffrom SDF to FS format, with an output filename of1000_chembl.fs
By using this FastSearch index, the speed of substructure and similarity searching is much improved. First of all, let’s do a substructure search:
Set up a conversion from FS to SMILES with
1000_chembl.fsas the input file. Tick the box for Output below only and Display in FirefoxEnter
Nc1ccc(N)cc1into the box Convert only if match SMARTS or mol in fileClick CONVERT
How does the speed of the substructure search compare to if you used
1000_chembl.sdfas the input file instead?
Next, let’s find the 5 most similar molecules to the same query. The Tanimoto coefficient of a path-based fingerprint is used as the measurement of similarity. This has a value from 0.0 to 1.0 (maximum similarity) and we will display the value below each molecule:
Set up the FS to SMILES conversion as before, and again enter
Nc1ccc(N)cc1into the box Convert only if match SMARTS or mol in fileEnter
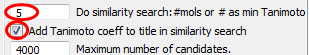
5into the box Do similarity search: #mols or # as min TanimotoTick the box Add Tanimoto coefficient to title in similarity search
Click CONVERT
Look at the 5 most similar molecules. Can you tell why they were regarded as similar to the query?