Molecular Similarity#
Substructure searching is a very powerful technique, but sometimes it misses answers for seemingly trivial differences. We saw this earlier with the following:
Query |
Target |
|---|---|
|
|
We’re looking for steroids… |
But we don’t find this one because of the double bond |
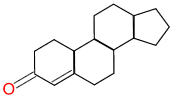
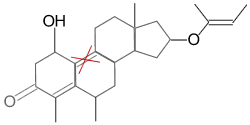
It is somewhat like searching for “221b Baker Street” and finding nothing because the database contains “221B Baker Street” and the system doesn’t consider “b” and “B” a match.
A good similarity search would find the target structure shown above, because even though it is not a substructure match, it is highly similar to our query.
There are many ways to measure similarity.
- 2D topology
The best-known and most widely used similarity metrics compare the two-dimensional topology, that is, they only use the molecule’s atoms and bonds without considering its shape.
Tanimoto similarity is perhaps the best known as it is easy to implement and fast to compute. An excellent summary of 2D similarity metrics can be found in section 5.3 of the Daylight Theory Manual.
- 3D configuration
One of the most important uses of similarity is in the discovery of new drugs, and a molecule’s shape is critical to it’s medicinal value (see QSAR).
3D similarity searches compare the configuration (also called the “conformation”) of a molecule to other molecules. The “electronic surface” of the molecule is the important bit - the part that can interact with other molecules. 3D searches compare the surfaces of two molecules, and how polarized or polarizable each bit of the surface is.
3D similarity searches are uncommon, for two reasons: It’s difficult and it’s slow. The difficulty comes from the complexity of molecular interactions - a molecule is not a fixed shape, but rather a dynamic object that changes according to its environment. And the slowness comes from the difficulty: To get better results, scientists employ more and more complex programs.
- Physical Properties
The above 2D and 3D similarity are based on the molecule’s structure. Another technique compares the properties - either computed or measured or both - and declares that molecules with many properties in common are likely to have similar structure. It is the idea of QSAR taken to the database.
- Clustering
“Clustering” is the process of differentiating a set of things into groups where each group has common features. Molecules can be clustered using a variety of techniques, such as common 2D and/or 3D features.
Note that clustering is not a similarity metric per se (the topic of this section), but it may use various similarity metrics when computing clusters. It is included here because it can be used as a “cheap substitute”. That is, when someone wants to find compounds similar to a known compound, you can show them the group (the cluster) to which the compound belongs. It allows you to pre-compute the clusters, spending lots of computational time up front, and then give answers very quickly.
Many cheminformatics databases have one or more similarity searches available.